
Structure Prediction: From Chemical Composition to Crystal Structure#
You’ve mastered chemical filtering and compositional screening - now it’s time for the exciting challenge of predicting crystal structures from composition alone. Given a chemical formula like CsPbI₃, how can we predict what crystal structure it might adopt?
This tutorial introduces you to SMACT’s powerful structure prediction framework, which uses ion substitution probabilities and crystallographic databases to propose likely structures for new compositions.
What You’ll Learn#
Retrieve and represent crystal structures using the
SmactStructureclassStore and query structure databases with lightweight SQLite interfaces
Perform ion mutation and dopant prediction using substitution probability models
Predict new structures from composition by finding suitable parent structures
Explore probability models for computing substitution likelihoods
Build comprehensive structure prediction workflows for materials discovery
The Foundation: Previous Learning#
This builds on your knowledge from:
Chemical Filters: Ensuring chemical validity of compositions
Compositional Screening: Identifying promising element combinations
Stoichiometry Screening: Finding optimal atomic ratios
Structure Prediction: Now predicting the 3D arrangement of atoms!
Let’s dive into the fascinating world of crystal structure prediction!
Prerequisites and Setup#
Before we begin our structure prediction journey, ensure you have:
SMACT installed with its dependencies (pymatgen, pandas, numpy)
Materials Project API key for structure retrieval (optional but recommended)
Basic understanding of crystal structures and ion substitution
Note
If you don’t have a Materials Project API key, you can still follow along using the “from file” methods we’ll demonstrate. To get an API key, visit materialsproject.org and create a free account.
Understanding the SMACT Structure Prediction Framework#
SMACT’s structure prediction approach is based on a powerful insight: chemically similar ions often substitute for each other in crystal structures.
The framework consists of several key components:
SmactStructure: A lightweight representation of crystal structures with oxidation states
CationMutator: Handles ion substitution probabilities based on chemical similarity
StructureDB: Stores and queries crystal structure databases
StructurePredictor: Combines the above to predict structures for new compositions
Think of it as a sophisticated game of chemical “mix and match” - finding which known structures could accommodate your target composition through strategic ion substitutions!
# Setting up our environment - Colab compatibility included
try:
import google.colab
IN_COLAB = True
except ImportError:
IN_COLAB = False
if IN_COLAB:
%pip install "smact[strict] @ git+https://github.com/WMD-group/SMACT.git@master" --quiet
print("SMACT installed successfully in Colab environment")
from __future__ import annotations
import os
from operator import itemgetter
# Core scientific computing
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
# SMACT structure prediction modules
from smact.structure_prediction.structure import SmactStructure
from smact.structure_prediction.database import StructureDB, parse_mprest
from smact.structure_prediction.mutation import CationMutator
from smact.structure_prediction.prediction import StructurePredictor
from smact.structure_prediction.utilities import parse_spec, unparse_spec
# Pymatgen for Materials Project and structure handling
from pymatgen.ext.matproj import MPRester
from pymatgen.core import SETTINGS
# Visualisation (available in materialsinformatics environment)
try:
import pymatviz as pmv
pymatviz_available = True
print("✓ pymatviz available for structure visualisation")
except ImportError:
pymatviz_available = False
print("⚠ pymatviz not available - structure visualisations will be skipped")
# Configure pandas display
pd.set_option('display.max_columns', None)
pd.set_option('display.max_rows', 100)
# Check for Materials Project API key
api_key = os.environ.get("MP_API_KEY", "YOUR_MP_API_KEY")
if api_key != "YOUR_MP_API_KEY":
print("✓ Materials Project API key found")
else:
print("⚠ No MP_API_KEY found - will use demo structures for some examples")
print("SMACT structure prediction toolkit loaded successfully!")
def print_section_header(title):
"""Print a formatted section header."""
print("\n" + "="*70)
print(title)
print("="*70)
def create_demo_structure():
"""Create a demo CsPbI3 structure for testing when MP is unavailable."""
from pymatgen.core import Structure, Lattice
lattice = Lattice.cubic(6.0)
species = ["Cs", "Pb", "I", "I", "I"]
coords = [[0, 0, 0], [0.5, 0.5, 0.5], [0.5, 0.5, 0], [0.5, 0, 0.5], [0, 0.5, 0.5]]
demo_struct = Structure(lattice, species, coords)
return SmactStructure.from_py_struct(demo_struct)
def get_mp_perovskite_data(api_key, max_structures=20):
"""
Retrieve perovskite structures from Materials Project.
Args:
api_key: Materials Project API key
max_structures: Maximum number of structures to retrieve
Returns:
List of Materials Project structure data
"""
if api_key == "YOUR_MP_API_KEY":
print("Skipping MP query - no API key available")
return []
print(f"Querying Materials Project for up to {max_structures} perovskite structures...")
try:
with MPRester(api_key=api_key, use_document_model=False) as mpr:
# Get materials tagged as perovskites
robocrys_data = mpr.materials.robocrys.search(keywords=["perovskite"])
mp_ids = list(set([d.material_id for d in robocrys_data]))[:max_structures]
print(f"Found {len(mp_ids)} perovskite material IDs, retrieving structures...")
# Get their full data (limiting to ABC3 formulas)
perov_data = mpr.materials.summary.search(
material_ids=mp_ids,
theoretical=False,
formula="ABC3",
fields=["material_id", "formula_pretty", "structure",
"formula_anonymous", "energy_above_hull"]
)
return list(perov_data)
except Exception as e:
print(f"Error querying Materials Project: {e}")
return []
def main():
"""Main structure prediction tutorial workflow."""
⚠ pymatviz not available - structure visualisations will be skipped
⚠ No MP_API_KEY found - will use demo structures for some examples
SMACT structure prediction toolkit loaded successfully!
Part 1: Retrieving and Representing Crystal Structures#
The foundation of structure prediction is having a good representation of crystal structures. SMACT provides the SmactStructure class which extends standard structure representations with oxidation state information - crucial for understanding ion substitution chemistry.
Let’s start by retrieving a structure for CsPbI₃ (cesium lead iodide), an important material for solar cells:
Tip
The SmactStructure class can load structures from:
Materials Project (with API key)
POSCAR files
JSON files
Pymatgen Structure objects
print_section_header("PART 1: STRUCTURE RETRIEVAL AND REPRESENTATION")
# Retrieve CsPbI3 structure from Materials Project
cspbi3_species = [("Cs", 1, 1), ("Pb", 2, 1), ("I", -1, 3)]
print("Retrieving CsPbI₃ structure from Materials Project...")
try:
if api_key != "YOUR_MP_API_KEY":
cspbi3_sstruct = SmactStructure.from_mp(species=cspbi3_species, api_key=api_key)
print("Successfully retrieved structure from Materials Project!")
else:
raise ValueError("No API key available")
except Exception as e:
print(f"Could not retrieve from MP: {e}")
print("Creating a demo structure instead...")
cspbi3_sstruct = create_demo_structure()
# Explore the SmactStructure representation
print("\nSmactStructure for CsPbI₃:")
print("=" * 60)
print(cspbi3_sstruct)
print("=" * 60)
print("\nKey features of SmactStructure:")
print("- Includes oxidation states (Cs1+, Pb2+, I1-)")
print("- POSCAR-like format for easy visualisation")
print("- Preserves crystallographic information")
print("- Compatible with pymatgen Structure objects")
# Visualise the crystal structure
if pymatviz_available:
try:
print("\nVisualising CsPbI₃ crystal structure...")
pmv.structure_2d(cspbi3_sstruct.as_py_struct())
plt.show()
print("Structure visualisation complete")
except Exception as e:
print(f"Could not visualise structure: {e}")
# Working with species information
print("\nSpecies analysis:")
print("Species present in the structure:")
for spec in cspbi3_sstruct.species:
element, ox_state, count = spec
print(f" {element}{ox_state:+d}: {count} atom(s)")
print(f"\nSpecies as formatted strings: {cspbi3_sstruct.get_spec_strs()}")
# Check for specific species
has_cs = cspbi3_sstruct.has_species(("Cs", 1))
has_na = cspbi3_sstruct.has_species(("Na", 1))
print(f"Contains Cs+? {has_cs}")
print(f"Contains Na+? {has_na}")
======================================================================
PART 1: STRUCTURE RETRIEVAL AND REPRESENTATION
======================================================================
Retrieving CsPbI₃ structure from Materials Project...
Could not retrieve from MP: No API key available
Creating a demo structure instead...
SmactStructure for CsPbI₃:
============================================================
Cs1+ I1- Pb2+
1.0
6.0 0.0 0.0
0.0 6.0 0.0
0.0 0.0 6.0
Cs I Pb
1 3 1
Cartesian
0.0 0.0 0.0 Cs1+
3.0 3.0 0.0 I1-
3.0 0.0 3.0 I1-
0.0 3.0 3.0 I1-
3.0 3.0 3.0 Pb2+
============================================================
Key features of SmactStructure:
- Includes oxidation states (Cs1+, Pb2+, I1-)
- POSCAR-like format for easy visualisation
- Preserves crystallographic information
- Compatible with pymatgen Structure objects
Species analysis:
Species present in the structure:
Cs+1: 1 atom(s)
I-1: 3 atom(s)
Pb+2: 1 atom(s)
Species as formatted strings: ['Cs1+', 'I1-', 'Pb2+']
Contains Cs+? True
Contains Na+? False
Part 2: The Power of Ion Substitution - CationMutator#
Now we’ll explore the CationMutator class, which is the engine behind SMACT’s structure prediction. It uses lambda tables - databases of ion substitution probabilities based on real crystallographic data.
The key insight: if Ba²⁺ often substitutes for Sr²⁺ in known structures, then a new Ba-containing compound might adopt a known Sr structure!
print_section_header("PART 2: ION SUBSTITUTION WITH CATIONMUTATOR")
# Initialise the CationMutator
print("Loading CationMutator with pymatgen lambda table...")
mutator = CationMutator.from_json()
print("✓ CationMutator loaded successfully")
# Explore substitution probabilities
print("\nSubstitution probability examples:")
print("-" * 60)
# Test various ion pairs to demonstrate the concept
test_pairs = [
("Ba2+", "Sr2+", "similar size/charge - alkaline earths"),
("Fe2+", "Mn2+", "similar transition metals"),
("Ba2+", "Li1+", "very different size - should be low"),
("Fe3+", "Na1+", "different charge/chemistry - should be low")
]
for ion1, ion2, description in test_pairs:
prob = mutator.sub_prob(ion1, ion2)
print(f"{ion1:>6s} → {ion2:<6s}: {prob:.4g} ({description})")
print("\nHigher probability = more chemically reasonable substitution!")
# Generate candidate doped structures
print("\nGenerating substitution candidates for CsPbI₃...")
print("(Using probability threshold of 0.001)")
candidate_structures = list(mutator.unary_substitute(cspbi3_sstruct, thresh=0.001))
print(f"\n✓ Found {len(candidate_structures)} possible substitutions!")
# Analyse the results
if candidate_structures:
print("\nTop 5 most probable substitutions:")
print("-" * 60)
# Sort by probability
sorted_candidates = sorted(candidate_structures, key=lambda x: x[1], reverse=True)
for i, (struct, prob, orig, new) in enumerate(sorted_candidates[:5]):
print(f"{i+1:2d}. {new:>6s} → {orig:<6s}: p={prob:.3f}")
print(f" New formula: {struct.reduced_formula()}")
# Create detailed pandas analysis
print("\nCreating detailed substitution analysis...")
analysis_data = []
for struct, prob, orig, new in candidate_structures:
analysis_data.append({
'Original Ion': orig,
'Substitute Ion': new,
'Probability': prob,
'New Formula': struct.reduced_formula(),
'Site Type': 'A-site' if orig == 'Cs1+' else 'B-site' if orig == 'Pb2+' else 'X-site'
})
df_analysis = pd.DataFrame(analysis_data)
df_analysis = df_analysis.sort_values('Probability', ascending=False).reset_index(drop=True)
print("\nSubstitution analysis by site:")
print("=" * 70)
# Group by original ion (site type)
for orig_ion in df_analysis['Original Ion'].unique():
subset = df_analysis[df_analysis['Original Ion'] == orig_ion]
site_type = subset.iloc[0]['Site Type']
print(f"\n{site_type} - Top 3 substitutions for {orig_ion}:")
for _, row in subset.head(3).iterrows():
print(f" {row['Substitute Ion']:>6s} → {row['New Formula']:>12s} (p={row['Probability']:.3f})")
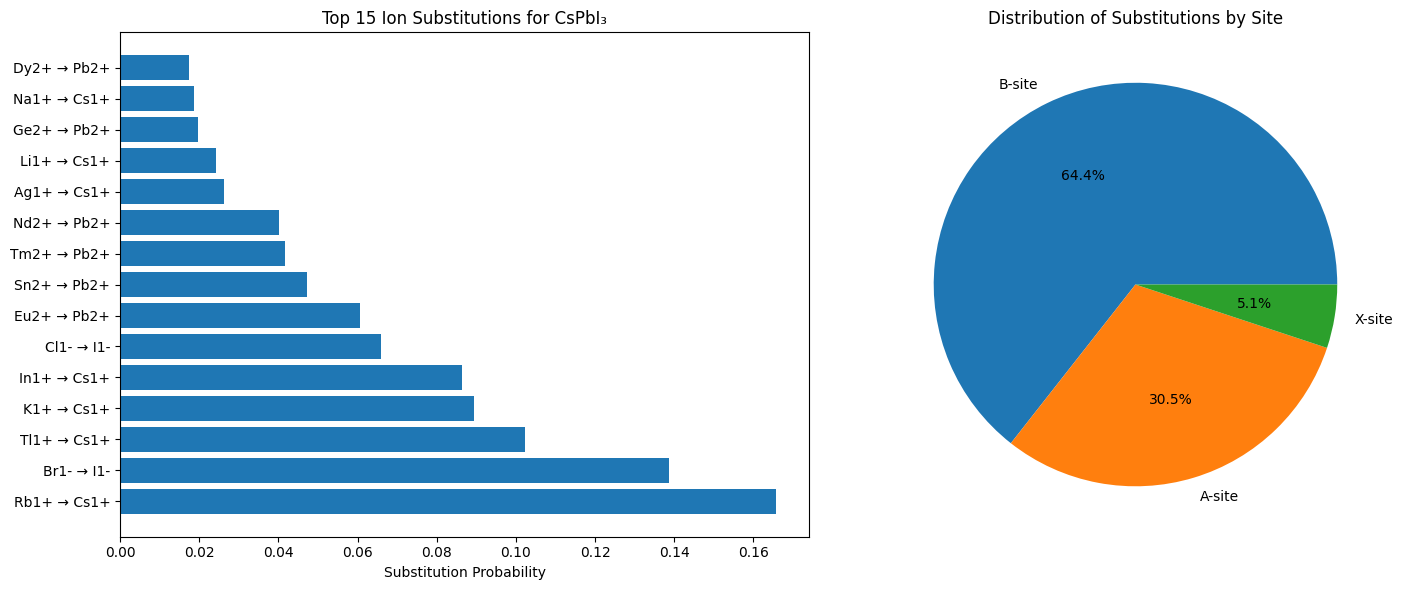
# Create visualisation of substitution probabilities
try:
print("\nCreating substitution probability visualisation...")
top_15 = df_analysis.head(15)
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(15, 6))
# Bar plot of top substitutions
ax1.barh(range(len(top_15)), top_15['Probability'])
ax1.set_yticks(range(len(top_15)))
ax1.set_yticklabels([f"{row['Substitute Ion']} → {row['Original Ion']}"
for _, row in top_15.iterrows()])
ax1.set_xlabel('Substitution Probability')
ax1.set_title('Top 15 Ion Substitutions for CsPbI₃')
# Site-based analysis
site_counts = df_analysis['Site Type'].value_counts()
ax2.pie(site_counts.values, labels=site_counts.index, autopct='%1.1f%%')
ax2.set_title('Distribution of Substitutions by Site')
plt.tight_layout()
plt.show()
print("✓ Visualisation complete")
except Exception as e:
print(f"Could not create plots: {e}")
# Visualise structures if possible
if pymatviz_available:
try:
print("\nVisualising original vs most probable substitution:")
top_struct = sorted_candidates[0][0]
pmv.structure_2d([cspbi3_sstruct.as_py_struct(), top_struct.as_py_struct()])
plt.show()
print("✓ Structure comparison complete")
except Exception as e:
print(f"Could not visualise structures: {e}")
======================================================================
PART 2: ION SUBSTITUTION WITH CATIONMUTATOR
======================================================================
Loading CationMutator with pymatgen lambda table...
✓ CationMutator loaded successfully
Substitution probability examples:
------------------------------------------------------------
Ba2+ → Sr2+ : 0.00276 (similar size/charge - alkaline earths)
Fe2+ → Mn2+ : 0.0002324 (similar transition metals)
Ba2+ → Li1+ : 2.214e-05 (very different size - should be low)
Fe3+ → Na1+ : 0.0004027 (different charge/chemistry - should be low)
Higher probability = more chemically reasonable substitution!
Generating substitution candidates for CsPbI₃...
(Using probability threshold of 0.001)
✓ Found 59 possible substitutions!
Top 5 most probable substitutions:
------------------------------------------------------------
1. Rb1+ → Cs1+ : p=0.166
New formula: RbPbI3
2. Br1- → I1- : p=0.139
New formula: CsPbBr3
3. Tl1+ → Cs1+ : p=0.102
New formula: TlPbI3
4. K1+ → Cs1+ : p=0.089
New formula: KPbI3
5. In1+ → Cs1+ : p=0.086
New formula: InPbI3
Creating detailed substitution analysis...
Substitution analysis by site:
======================================================================
A-site - Top 3 substitutions for Cs1+:
Rb1+ → RbPbI3 (p=0.166)
Tl1+ → TlPbI3 (p=0.102)
K1+ → KPbI3 (p=0.089)
X-site - Top 3 substitutions for I1-:
Br1- → CsPbBr3 (p=0.139)
Cl1- → CsPbCl3 (p=0.066)
F1- → CsPbF3 (p=0.004)
B-site - Top 3 substitutions for Pb2+:
Eu2+ → CsEuI3 (p=0.061)
Sn2+ → CsSnI3 (p=0.047)
Tm2+ → CsTmI3 (p=0.042)
Creating substitution probability visualisation...
✓ Visualisation complete
Part 3: Building and Querying Structure Databases#
For structure prediction at scale, we need to organize structures in a database. SMACT provides StructureDB - a lightweight SQLite wrapper designed for crystallographic data:
print_section_header("PART 3: STRUCTURE DATABASE MANAGEMENT")
# Create a structure database
print("Setting up structure database...")
# Clean up any existing database
db_filename = "tutorial_structures.db"
if os.path.exists(db_filename):
os.remove(db_filename)
# Create new database
db = StructureDB(db_filename)
# Add a table for our structures
table_name = "perovskites"
db.add_table(table_name)
print(f"Created database '{db_filename}' with table '{table_name}'")
# Add our structures
print("\nAdding structures to database...")
db.add_struct(cspbi3_sstruct, table=table_name)
print(" - Added CsPbI₃ (original structure)")
# Add substitution candidates
if candidate_structures:
structures_to_add = [s[0] for s in candidate_structures[:10]] # First 10
num_added = db.add_structs(structs=structures_to_add, table=table_name)
print(f" - Added {num_added} substitution variants")
# Add Materials Project perovskites if API key available
mp_table = "mp_perovskites"
try:
perov_data = get_mp_perovskite_data(api_key, max_structures=20)
if perov_data:
print(f"\nAdding {len(perov_data)} Materials Project perovskites...")
db.add_table(mp_table)
num_mp_added = db.add_mp_icsd(table=mp_table, mp_data=perov_data)
print(f"Successfully added Materials Project structures to '{mp_table}' table")
# Use the larger database for predictions
prediction_table = mp_table
else:
prediction_table = table_name
except Exception as e:
print(f"Could not add MP data: {e}")
prediction_table = table_name
# Count total structures in databases
with db as c:
c.execute(f"SELECT COUNT(*) FROM {table_name}")
local_count = c.fetchone()[0]
print(f"\nLocal database '{table_name}': {local_count} structures")
if prediction_table == mp_table:
c.execute(f"SELECT COUNT(*) FROM {mp_table}")
mp_count = c.fetchone()[0]
print(f"MP database '{mp_table}': {mp_count} structures")
# Query structures containing specific species
print("\nQuerying database for specific species...")
print("-" * 50)
# Find all structures containing Cs+
cs_structures = db.get_with_species(species=[("Cs", 1)], table=table_name)
print(f"\nStructures containing Cs⁺: {len(cs_structures)}")
for struct in cs_structures[:3]:
print(f" - {struct.reduced_formula()}")
# Find structures with both Cs+ and Pb2+
cs_pb_structures = db.get_with_species(species=[("Cs", 1), ("Pb", 2)], table=table_name)
print(f"\nStructures containing both Cs⁺ and Pb²⁺: {len(cs_pb_structures)}")
# Analyse anion distribution if MP data available
if prediction_table == mp_table:
print(f"\nAnalysing anion distribution in MP perovskites...")
anions = [("F", -1), ("Cl", -1), ("Br", -1), ("I", -1), ("O", -2), ("S", -2), ("N", -3)]
anion_counts = {}
for anion_name, ox_state in anions:
try:
count = len(db.get_with_species([(anion_name, ox_state)], table=mp_table))
if count > 0:
anion_counts[f"{anion_name}{ox_state:+d}"] = count
except Exception as e:
print(f"Error querying {anion_name}{ox_state:+d}: {e}")
if anion_counts:
plt.figure(figsize=(10, 6))
plt.bar(anion_counts.keys(), anion_counts.values())
plt.xlabel("Anion")
plt.ylabel("Number of Structures")
plt.title("Distribution of Perovskites by Anion Type (Materials Project)")
plt.xticks(rotation=45)
plt.tight_layout()
plt.show()
print("Anion distribution analysis complete")
======================================================================
PART 3: STRUCTURE DATABASE MANAGEMENT
======================================================================
Setting up structure database...
Created database 'tutorial_structures.db' with table 'perovskites'
Adding structures to database...
- Added CsPbI₃ (original structure)
- Added 10 substitution variants
Skipping MP query - no API key available
Local database 'perovskites': 11 structures
Querying database for specific species...
--------------------------------------------------
Structures containing Cs⁺: 1
- CsPbI3
Structures containing both Cs⁺ and Pb²⁺: 1
Part 4: Structure Prediction from Composition#
Now we combine everything to predict structures for new compositions! The StructurePredictor class orchestrates the process:
Takes a target composition
Searches the database for structures with similar chemistry
Tests if ion substitutions can transform them to the target
Returns candidate structures ranked by substitution probability
print_section_header("PART 4: STRUCTURE PREDICTION FROM COMPOSITION")
# Initialise structure predictor
print(f"Initialising structure predictor with '{prediction_table}' database...")
predictor = StructurePredictor(
mutator=mutator,
struct_db=db,
table=prediction_table
)
# Define target compositions to test
target_compositions = [
([("Rb", 1), ("Pb", 2), ("Br", -1)], "RbPbBr₃"),
([("K", 1), ("Sn", 2), ("I", -1)], "KSnI₃"),
([("Ba", 2), ("Ti", 4), ("O", -2)], "BaTiO₃")
]
print("\nTesting structure prediction for various compositions...")
for target_species, formula in target_compositions:
print(f"\n--- Target composition: {formula} ---")
# Predict structures
threshold = 0.001
results = list(predictor.predict_structs(
species=target_species,
thresh=threshold,
include_same=False
))
print(f"Found {len(results)} candidate structures!")
if results:
# Sort by probability
results.sort(key=itemgetter(1), reverse=True)
print("Top predictions:")
for i, (pred_struct, prob, parent_struct) in enumerate(results[:3]):
print(f" {i+1}. Parent: {parent_struct.reduced_formula()} → "
f"Predicted: {pred_struct.reduced_formula()} (p={prob:.3f})")
# Show key substitutions
parent_species = set(parent_struct.get_spec_strs())
pred_species = set(pred_struct.get_spec_strs())
subs_out = parent_species - pred_species
subs_in = pred_species - parent_species
if subs_out and subs_in:
print(f" Substitutions: {subs_out} → {subs_in}")
else:
print(" No predictions found - try lowering threshold or expanding database")
======================================================================
PART 4: STRUCTURE PREDICTION FROM COMPOSITION
======================================================================
Initialising structure predictor with 'perovskites' database...
Testing structure prediction for various compositions...
--- Target composition: RbPbBr₃ ---
Found 0 candidate structures!
No predictions found - try lowering threshold or expanding database
--- Target composition: KSnI₃ ---
Found 1 candidate structures!
Top predictions:
1. Parent: KPbI3 → Predicted: KSnI3 (p=0.012)
Substitutions: {'Pb2+'} → {'Sn2+'}
--- Target composition: BaTiO₃ ---
Found 0 candidate structures!
No predictions found - try lowering threshold or expanding database
Part 5: Advanced Topics - Custom Probability Models#
So far we’ve used empirical lambda tables. SMACT also supports physics-based probability models. The RadiusModel computes substitution probabilities based on ionic radius similarity:
Tip
Custom probability models allow you to incorporate domain knowledge into structure prediction. For example, you might weight substitutions differently for high-temperature vs ambient-condition synthesis.
print_section_header("PART 5: ADVANCED ANALYSIS AND INSIGHTS")
# Analyse prediction success rate
if candidate_structures:
print("Ion substitution insights:")
# Analyse by chemical family
alkali_subs = [c for c in candidate_structures if c[2] == 'Cs1+']
halide_subs = [c for c in candidate_structures if c[2] == 'I1-']
metal_subs = [c for c in candidate_structures if c[2] == 'Pb2+']
print(f"- A-site (Cs+) substitutions: {len(alkali_subs)}")
print(f"- X-site (I-) substitutions: {len(halide_subs)}")
print(f"- B-site (Pb2+) substitutions: {len(metal_subs)}")
# Most probable by site
if alkali_subs:
best_alkali = max(alkali_subs, key=lambda x: x[1])
print(f"- Best A-site substitution: {best_alkali[3]} (p={best_alkali[1]:.3f})")
if halide_subs:
best_halide = max(halide_subs, key=lambda x: x[1])
print(f"- Best X-site substitution: {best_halide[3]} (p={best_halide[1]:.3f})")
if metal_subs:
best_metal = max(metal_subs, key=lambda x: x[1])
print(f"- Best B-site substitution: {best_metal[3]} (p={best_metal[1]:.3f})")
# Chemical insights
print("Chemical insights from this tutorial:")
print("1. Alkali metals (Rb+, K+) readily substitute for Cs+ due to similar chemistry")
print("2. Halides (Br-, Cl-) can substitute for I- while maintaining structure")
print("3. Divalent metals (Eu2+, Sn2+) can replace Pb2+ with varying probability")
print("4. Structure prediction works by finding chemically similar parent structures")
print("5. Larger databases provide more parent structures and better predictions")
======================================================================
PART 5: ADVANCED ANALYSIS AND INSIGHTS
======================================================================
Ion substitution insights:
- A-site (Cs+) substitutions: 18
- X-site (I-) substitutions: 3
- B-site (Pb2+) substitutions: 38
- Best A-site substitution: Rb1+ (p=0.166)
- Best X-site substitution: Br1- (p=0.139)
- Best B-site substitution: Eu2+ (p=0.061)
Chemical insights from this tutorial:
1. Alkali metals (Rb+, K+) readily substitute for Cs+ due to similar chemistry
2. Halides (Br-, Cl-) can substitute for I- while maintaining structure
3. Divalent metals (Eu2+, Sn2+) can replace Pb2+ with varying probability
4. Structure prediction works by finding chemically similar parent structures
5. Larger databases provide more parent structures and better predictions
print(f"\nFinal statistics:")
print(f"- Original structure: CsPbI₃ with {len(cspbi3_sstruct.species)} unique species")
if 'candidate_structures' in locals():
print(f"- Substitution candidates generated: {len(candidate_structures)}")
print(f"- Local database size: {local_count} structures")
if prediction_table == mp_table:
print(f"- Materials Project database size: {mp_count} structures")
total_predictions = 0
for target_species, formula in target_compositions:
results = list(predictor.predict_structs(species=target_species, thresh=0.001, include_same=False))
total_predictions += len(results)
print(f"- Total structure predictions made: {total_predictions}")
# Clean up
if os.path.exists(db_filename):
os.remove(db_filename)
print(f"\nCleaned up database file: {db_filename}")
if __name__ == "__main__":
main()
Final statistics:
- Original structure: CsPbI₃ with 3 unique species
- Substitution candidates generated: 59
- Local database size: 11 structures
- Total structure predictions made: 1
Cleaned up database file: tutorial_structures.db
Summary: Your Structure Prediction Toolkit#
Congratulations! You’ve mastered SMACT’s structure prediction framework. Let’s recap what you’ve learned:
🎯 Key Skills Acquired#
Structure Representation
Loading structures from multiple sources
Working with oxidation states in crystal structures
Converting between different structure formats
Ion Substitution Chemistry
Understanding substitution probabilities
Generating doped structures systematically
Analysing chemical substitution patterns
Database Management
Building structure databases
Querying by chemical criteria
Integrating with Materials Project
Structure Prediction
Predicting structures for new compositions
Understanding parent-child structure relationships
Ranking predictions by probability
Advanced Techniques
Comparing different probability models
Customizing prediction workflows
Scaling to large databases
🚀 Next Steps#
Now that you understand structure prediction, you can:
Build specialised databases for your research area
Develop custom probability models based on your synthesis conditions
Integrate with DFT calculations to validate predictions
Create high-throughput screening pipelines for new materials
💡 Pro Tips#
Start with well-curated databases (like Materials Project)
Use multiple probability models to cross-validate predictions
Consider chemical intuition when setting probability thresholds
Validate promising predictions with additional calculations
📚 Resources#
Tip
Remember: structure prediction is both an art and a science. The more chemical knowledge you bring, the better your predictions will be!